
De acuerdo con la Dra. Fabiola Mariño, Gerente Médico de Enfermedades Raras de Pfizer México, este padecimiento fue descrito por primera vez en 1882 por el médico francés Philippe Charles Ernest Gaucher y pertenece al grupo de enfermedades de depósito lisosomal.
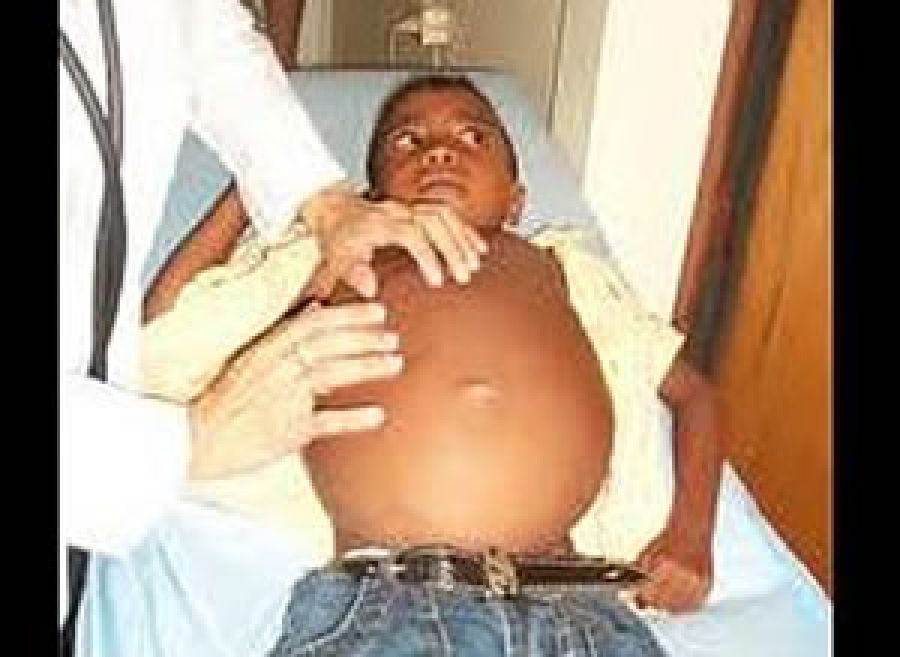
Se trata, dice, de un trastorno metabólico hereditario, poco frecuente, causado por la ausencia o deficiencia de la enzima glucocerebrosidasa (participante en la degradación de lípidos), lo que conlleva la acumulación de depósitos grasos en las células de algunos órganos como hígado, bazo, huesos y médula ósea, lo que interfiere con su adecuado funcionamiento. “La acumulación de células dañinas en la médula ósea puede mermar la producción de glóbulos rojos, leucocitos y plaquetas”, señala la especialista.
Comenta que, para que se presente esta enfermedad, deben coincidir dos portadores (padres) con un gen anormal, que se transmite a los hijos, quienes pueden manifestarla en cualquier etapa afectando considerablemente la salud y calidad de vida de quien la presenta. De hecho, puede ser mortal si aparece a edad temprana o no se atiende oportunamente.
“Dado que los lisosomas –vesículas encargadas de la digestión intracelular– están presentes en la mayoría de las células, la enfermedad de Gaucher afecta de forma variable al cerebro y las funciones cognitivas”, comenta.
Si en una familia se ha identificado un integrante que padece enfermedad de Gaucher, los padres y hermanos deben ser referidos al médico genetista para recibir asesoramiento genético.
La Dra. Mariño explica que, si bien tradicionalmente se ha clasificado en tres formas clínicas, basándose en la ausencia (tipo I) o presencia (tipos II y III) de un padecimiento en el sistema nervioso, existen cinco tipos de la enfermedad de Gaucher, los cuales se clasifican con base en sus manifestaciones clínicas[3]:
· Tipo I. Es la más común (aproximadamente 95% de los casos), afectando tanto a niños como adultos.
· Tipo II. Generalmente comienza durante la lactancia. Se presenta en el 1% de los casos y puede llevar a una muerte rápida y temprana.
· Tipo III. Las personas con esta clasificación (el 4% de los casos) pueden vivir hasta la edad adulta.[4]
· Perinatal Letal. Es la forma más grave y sus complicaciones pueden comenzar antes del nacimiento o en primera infancia.[5]
· Cardiovascular. Afecta principalmente al corazón.[6]
Para la enfermedad de Gaucher se requiere un diagnóstico adecuado, así como la participación de un equipo multidisciplinario que permita la atención integral del paciente.
“Si bien se han producido avances significativos en el diagnóstico y la terapia, existe una necesidad continua de ampliar el conocimiento acerca de la enfermedad”, finaliza la especialista.
Redacción MD
--------------------
Referencias:
[1] Franco-Ornelas, Sergio, et al.. (2012). Enfermedad de Gaucher: hallazgos clínicos en 11 niños. 22 de septiembre de 2017, de Instituto Mexicano del Seguro Social Sitio web: http://www.redalyc.org/html/4577/457745493020/
[2] The Gaucher Registry. (2017). Disease Classification. 28 de septiembre de 2017, de The Gaucher Registry Sitio web: https://www.gauchercare.com/healthcare/information/DiseaseClassification.aspx
[3] Martínez Álvarez, Iván, et al.. (2011). Enfermedad de Gaucher. Reporte de un caso y revisión de la bibliografía. 22 de septiembre de 2017, de Medicina Interna de México Sitio web: http://www.medigraphic.com/pdfs/medintmex/mim-2011/mim112o.pdf
[4] Biblioteca Nacional de Medicina de los EE.UU.. (2017). Enfermedad de Gaucher. 24 de septiembre de 2017, de Biblioteca Nacional de Medicina de los EE.UU. Sitio web: https://medlineplus.gov/spanish/ency/article/000564.htm
[5] Infogen. (2017). Enfermedad de Gaucher. 22 de septiembre de 2017, de Infogen Sitio web: http://infogen.org.mx/enfermedad-de-gaucher-sindrome-de-gaucher-2/
[6] Ídem.